

 021-51111890
021-51111890 购物车()
购物车()
 sales@molnova.cn
sales@molnova.cn





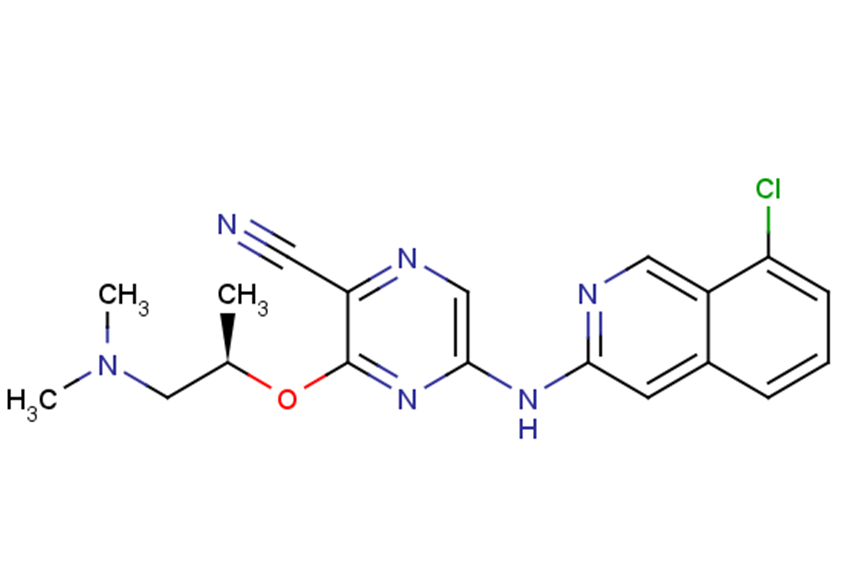
SAR-020106
CAS No. 1184843-57-9
SAR-020106 ( —— )
产品货号. M22629 CAS No. 1184843-57-9
SAR-020106 是一种有效的、ATP 竞争性的、选择性的 CHK1 抑制剂,对分离的人酶的 IC50 为 13.3 nmol/L。SAR-020106 是一种 ATP 竞争性的、有效的、选择性的 CHK1 抑制剂,IC50 (50)分离的人酶的浓度为 13.3 nmol/L。该化合物可消除依托泊苷诱导的 G(2) 阻滞,在 HT29 细胞中 IC(50) 为 55 nmol/L,并在多种结肠肿瘤系中显着增强吉西他滨和 SN38 的细胞杀伤力 3.0 至 29 倍体外并以 p53 依赖性方式。
纯度: >98% (HPLC)
 COA
Datasheet
HNMR
HPLC
MSDS
Handing Instructions
COA
Datasheet
HNMR
HPLC
MSDS
Handing Instructions
| 规格 | 价格/人民币 | 库存 | 数量 |
| 5MG | ¥931 | 有现货 |


|
| 10MG | ¥1492 | 有现货 |
|
| 25MG | ¥3143 | 有现货 |
|
| 50MG | ¥4483 | 有现货 |
|
| 100MG | ¥6021 | 有现货 |
|
| 500MG | 获取报价 | 有现货 |
|
| 1G | 获取报价 | 有现货 |
|
| 1 mL x 10 mM in DMSO | ¥1121 | 有现货 |
|
生物学信息
-
产品名称SAR-020106
-
注意事项本公司产品仅用于科研实验,不得用于人体或动物的临床与诊断
-
产品简述SAR-020106 是一种有效的、ATP 竞争性的、选择性的 CHK1 抑制剂,对分离的人酶的 IC50 为 13.3 nmol/L。SAR-020106 是一种 ATP 竞争性的、有效的、选择性的 CHK1 抑制剂,IC50 (50)分离的人酶的浓度为 13.3 nmol/L。该化合物可消除依托泊苷诱导的 G(2) 阻滞,在 HT29 细胞中 IC(50) 为 55 nmol/L,并在多种结肠肿瘤系中显着增强吉西他滨和 SN38 的细胞杀伤力 3.0 至 29 倍体外并以 p53 依赖性方式。
-
产品描述SAR-020106 is a potent, ATP-competitive, and selective CHK1 inhibitor with an IC50 of 13.3 nmol/L on the isolated human enzyme.SAR-020106 is an ATP-competitive, potent, and selective CHK1 inhibitor with an IC(50) of 13.3 nmol/L on the isolated human enzyme. This compound abrogates an etoposide-induced G(2) arrest with an IC(50) of 55 nmol/L in HT29 cells, and significantly enhances the cell killing of gemcitabine and SN38 by 3.0- to 29-fold in several colon tumor lines in vitro and in a p53-dependent fashion. Biomarker studies have shown that SAR-020106 inhibits cytotoxic drug-induced autophosphorylation of CHK1 at S296 and blocks the phosphorylation of CDK1 at Y15 in a dose-dependent fashion both in vitro and in vivo. Cytotoxic drug combinations were associated with increased gammaH2AX and poly ADP ribose polymerase cleavage consistent with the SAR-020106-enhanced DNA damage and tumor cell death. Irinotecan and gemcitabine antitumor activity was enhanced by SAR-020106 in vivo with minimal toxicity. SAR-020106 represents a novel class of CHK1 inhibitors that can enhance antitumor activity with selected anticancer drugs in vivo and may therefore have clinical utility.SAR-020106 potentiated the efficacies of irinotecan and gemcitabine in SW620 human colon carcinoma xenografts in nude mice.
-
体外实验SAR-020106 (0.1-1 μM; 23 hours) abrogates an Etoposide-induced S and G2 arrest.SAR-020106 is capable of abrogating Etoposide-induced cell cycle arrest with an IC50 of 55 nM and 91 nM in HT29 and SW620 cells, respectively. SAR-020106 is relatively nontoxic with a GI50 of 0.48 μM in HT29 and 2 μM in SW620, resulting in an activity index of 8.7 and 22, respectively. SAR-020106 inhibits cytotoxic drug-induced autophosphorylation of CHK1 at S296 and blocks the phosphorylation of CDK1 at Y15 in a dose-dependent fashion.
-
体内实验SAR-020106 (40 mg/kg; i.p.; administered on days 0, 1, 7, 8, 14, and 15) in combination with Irinotecan potentiates the antitumor activity in SW620 xenografts. Animal Model:Nude mice bearing SW620 xenograft tumors Dosage:40 mg/kg Administration:I.p.; administered on days 0, 1, 7, 8, 14, and 15 Result:There was a clear decrease in tumor growth associated with the combination with tumors reaching 300% by 12.5 days.
-
同义词——
-
通路Angiogenesis
-
靶点Chk
-
受体CHK1
-
研究领域——
-
适应症——
化学信息
-
CAS Number1184843-57-9
-
分子量382.85
-
分子式C19H19ClN6O
-
纯度>98% (HPLC)
-
溶解度In Vitro:?DMSO : 5 mg/mL (13.06 mM)
-
SMILESC[C@H](CN(C)C)Oc1nc(Nc2cc3cccc(Cl)c3cn2)cnc1C#N
-
化学全称——
运输与储存
-
储存条件(-20℃)
-
运输条件With Ice Pack
-
稳定性≥ 2 years
参考文献
1. Walton M I , Eve P D , Hayes A , et al. The Preclinical Pharmacology and Therapeutic Activity of the Novel CHK1 Inhibitor SAR-020106[J]. Molecular Cancer Therapeutics, 2010, 9(1):89-100.
产品手册




关联产品
-
XL-844
一种有效、特异性、口服的 Chk1 和 Chk2 ATP 竞争性抑制剂,Ki 分别为 2.2 nM 和 0.07 nM。
-
BAY-1816032
BAY-1816032 (BAY1816032) 是一种高效、选择性、口服活性的 BUB1 有丝分裂检查点丝氨酸/苏氨酸激酶,IC50 为 7 nM。
-
VRX-0466617
一种有效的选择性 Chk2 抑制剂,Ki/IC50 为 11 nM/120 nM;对 Chk1 (>100 uM) 无抑制作用。


